Graph & Geometric ML in 2024: Where We Are and What’s Next

Structural Biology (Molecules & Proteins)
Dominique Beaini (Valence), Joey Bose (Mila & Dreamfold), Michael Bronstein (Oxford), Bruno Correia (EPFL), Michael Galkin (Intel), Kexin Huang (Stanford), Chaitanya Joshi (Cambridge), Andreas Loukas (Genentech), Luca Naef (VantAI), Hannes Stärk (MIT), Minkai Xu (Stanford)
Structural biology was definitely at the forefront of Geometric Deep Learning in 2023.
Following the 2020 discovery of halicin as a potential new antibiotic, in 2023, two new antibiotics were discovered with the help of GNNs! First, it is abaucin (by McMaster and MIT), which targets a stubborn pathogen resistant to many drugs. Second, MIT and Harvard researchers discovered a new structural class of antibiotics where the screening process was supported by ChemProp, a suite of GNNs for molecular property prediction. We also observe a convergence of ML and experimental techniques (“lab-in the-loop”) in the recent work on autonomous molecular discovery (a trend we will also see in the Materials Design in the following sections).
Flow Matching has been one of the biggest generative ML trends of 2023, allowing for faster sampling and deterministic sampling trajectories compared to diffusion models. The most prominent examples of Flow Matching models we have seen in the biological applications are FoldFlow (Bose, Akhound-Sadegh, et al.) for protein backbone generation, FlowSite (Stärk et al.) for protein binding site design, and EquiFM (Song, Gong, et al.) for molecule generation.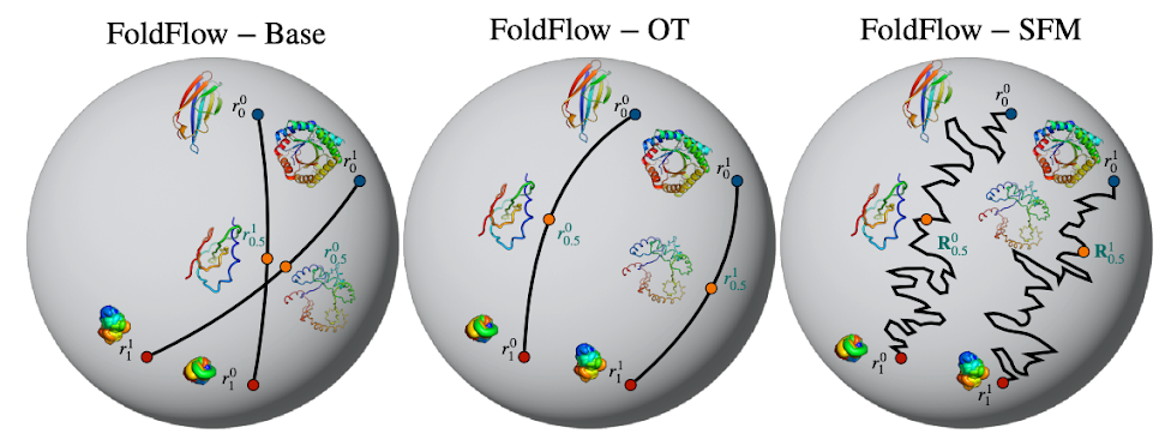 Conditional probability paths learned by different versions of FoldFlow, visualizing the rotation trajectory of a single residue by the action of SO(3) on its homogeneous space 𝕊². Figure source: Bose, Akhound-Sadegh, et al.
Conditional probability paths learned by different versions of FoldFlow, visualizing the rotation trajectory of a single residue by the action of SO(3) on its homogeneous space 𝕊². Figure source: Bose, Akhound-Sadegh, et al.
Efficient Flow Matching on complex geometries with necessary equivariances became possible thanks to a handful of theory papers including Riemannian Flow Matching (Chen and Lipman), Minibatch Optimal Transport (Tong et al), and Simulation-Free Schrödinger bridges (Tong, Malkin, Fatras, et al). A great resource to learn Flow Matching with code examples and notebooks is the TorchCFM repo on GitHub as well as talks by Yaron Lipman, Joey Bose, Hannes Stärk, and Alex Tong.
Diffusion models nevertheless continue to be the main workhorse of generative modeling in structural biology. In 2023, we saw several landmark works: FrameDiff (Yim, Trippe, De Bortoli, Mathieu, et al) for protein backbone generation, EvoDiff (Alamdari et al) for generating protein sequences with discrete diffusion, AbDiffuser (Martinkus et al) for full-atom antibody design with frame averaging and discrete diffusion (and with successful wet lab experiments), DiffMaSIF (Sverrison, Akdel, et al) and DiffDock-PP (Ketata, Laue, Mammadov, Stärk, et al) for protein-protein docking, DiffPack (Zhang, Zhang, et al) for side-chain packing, and the Baker Lab published the RFDiffusion all-atom version (Krishna, Wang, Ahern, et al). Among latent diffusion model (like Stable Diffusion in image generation applications), GeoLDM (Xu et al) was the first for 3D molecule conformations, followed by OmniProt for protein sequence-structure generation.FrameDiff: parameterization of the backbone frame with rotation, translation, and torsion angle for the oxygen atom. Figure Source: Yim, Trippe, De Bortoli, Mathieu, et al
Finally, Google DeepMind and Isomorphic Labs announced AlphaFold 2.3 — the latest iteration is significantly improving upon the baselines in 3 tasks: docking benchmarks (almost 2× better than DiffDock on the new PoseBusters benchmark), protein-nucleic acid interactions, and antibody-antigen prediction.
Chaitanya Joshi (Cambridge)
💡There have been two emerging trends for biomolecular modeling and design that I am very excited about in 2023:
1️⃣ Going from protein structure prediction to conformational ensemble generation. There were several interesting approaches to the problem, including AlphaFold with MSA clustering, idpGAN, Distributional Graphormer (a diffusion model), and AlphaFold Meets Flow Matching for Generating Protein Ensembles.
2️⃣ Modelling of biomolecular complexes and design of biomolecular interactions among proteins + X: RFdiffusion all-atom and Ligand MPNN, both from the Baker Lab, are representative examples of the trend towards designing interactions. The new in-development AlphaFold report claims that a unified structure prediction model can outperform or match specialised models across solo protein and protein complex structure prediction as well as protein-ligand and protein-nucleic acid co-folding.
“However, for all the exciting methodology development in biomolecular modelling and design, perhaps the biggest lesson for the ML community this year should be to focus more on meaningful in-silico evaluation and, if possible, experimental validation.” — Chaitanya Joshi (Cambridge)
1️⃣ In early 2023, Guolin Ke’s team at DP Technology released two excellent re-evaluation papers highlighting how we may have been largely overestimating the performance of prominent geometric deep learning-based methods for molecular conformation generation and docking w.r.t. traditional baselines.
2️⃣ PoseCheck and PoseBusters shed further light on the failure modes of current molecular generation and docking methods. Critically, generated molecules and their 3D poses are often ‘nonphysical’ and contain steric clashes, hydrogen placement issues, and high strain energies.
3️⃣ Very few papers attempt any experimental validation of new ML ideas. Perhaps collaborating with a wet lab is challenging for those focussed on new methodology development, but I hope that us ML-ers, as a community, will at least be a lot more cautious about the in-silico evaluation metrics we are constantly pushing as we create new models.
Hannes Stärk (MIT)
💡I am reading quite some hype here about Flow Matching, stochastic interpolants, and Rectified Flows (I will call them “Bridge Matching,” or “BM”). I do not think there is much value in just replacing diffusion models with BM in all the existing applications. For pure generative modeling, the main BM advantage is simplicity.
I think we should instead be excited about BM for the new capabilities it unlocks. For example, training bridges between arbitrary distributions in a simulation-free manner (what are the best applications for this? I basically only saw retrosynthesis so far.) or solving OT problems as in DSBM that does so for fluid flow downscaling. Maybe a lot of tools emerged in 2023 (also let us mention BM with multiple marginals), and in 2024, the community will make good use of them?
Joey Bose (Mila & Dreamfold)
💡 This year we have really seen the rise of geometric generative models from theory to practice. A few standouts for me include Riemannian Flow Matching — in general any paper by Ricky Chen and Yaron Lipman on these topics is a must-read — and FrameDiff from Yim et. al which introduced a lot of the important machinery for protein backbone generation. Of course, standing on the shoulders of both RFM and FrameDIff, we built FoldFlow, a cooler flow-matching approach to protein generative models.
“Looking ahead, I foresee a lot more flow matching-based approaches coming into use. They are better for proteins and longer sequences and can start from any source distribution.” — Joey Bose (Mila & Dreamfold)
🔮 Moreover, I suspect we will soon see multi-modal generative models in this space, such as discrete + continuous models and also conditional models in the same vein as text-conditioned diffusion models for images. Perhaps, we might even see latent generative models here given that they scale so well!
Minkai Xu (Stanford)
“This year, the community has further pushed forward the geometric generative models for 3D molecular generation in many perspectives.” — Minkai Xu (Stanford)
Flow matching: Ricky and Yaron proposed the Flow Matching method as an alternative to the widely used diffusion models, and EquiFM (Song et al and Klein et al) realize the variant for 3D molecule generation by parameterizing the flow dynamics with equivariant GNNs. In the meantime, FrameFlow and FoldFlow construct FM models for protein generation.
🔮Moving forward similar to vision and text domain, people begin to explore generation in the lower-dimensional latent space instead of the complex original data space (latent generative models). GeoLDM (Xu et al) proposed the first latent diffusion model (like Stable Diffusion in CV) for 3D molecule generation, while Fu et al enjoys similar modeling formulation for large protein generation.
A Structural Biologist’s Perspective
Bruno Correia (EPFL)
“Current generative models still create “garbage” outputs that violate many of the physical and chemical properties that molecules are known to have. The advantage of current generative models is, of course, their speed which affords them the possibility of generating many samples, which then brings to front and center the ability to filter the best generated samples, which in the case of protein design has benefited immensely from the transformative development of AlphaFold2.” — Bruno Correia (EPFL)
➡️ The next challenge to the community will perhaps be how to infuse generative models with meaningful physical and chemical priors to enhance sampling performance and generalization. Interestingly, we have not seen the same remarkable advances (experimentally validated) in applications to small molecule design, which we hope to see during 2024.
➡️ The rise of multimodal models. Generally in biological-related tasks data sparsity is a given and as such strategies to extract the most signal out of the data are essential. One way to try to overcome such limitations is to improve the expressiveness of the data representations and maybe this way obtain more performant neural networks. Likely in the short term, we will be able to explore architectures that encompass several types of representations of the objects of interest and harness the best predictions for the evermore complex tasks we are facing as progressively more of the basic problems get solved. This notion of multimodality is of course intimately related to the overall aim of having models with stronger priors, that in a generative context, honour fundamental constraints of the objects of interest.
➡️ The models that know everything. As the power of machine learning models improves we clearly tend to request a more multi-objective optimization when it comes to attempting to solve real life problems. Taking as an example small molecule generation, thinking from a biochemical perspective the drug design problem starts by having a target to which a small molecule binds, therefore one of the first and most important constraints is that the generative process ought to be conditioned to the protein pocket. However, such a constraint may not be enough to create real small molecules as many of such chemicals are simply impossible or very hard to synthesize, and, therefore, a model that has notions of chemical synthesizability and can integrate such constraints in the search space would be much more useful.
➡️ From chemotype to phenotype. On the grounds of data representation, atomic graph structures together with vector embeddings have reached remarkable results, particularly in the search for new antibiotics. Making accurate predictions of which chemical structures have antimicrobial activity, broadly speaking, is an exercise of phenotype prediction from chemical structure. Due to the simplicity of the approaches used and the impressive results obtained, one would expect that more sophisticated data representations on the molecule end and perhaps together also with richer phenotype assignment could give critical contributions to such an important problem in drug development.
Industrial perspective
Luca Naef (VantAI)
🔥What are the biggest advancements in the field you noticed in 2023?
1️⃣ Increasing multi-modality & modularity — as shown by the emergence of initial co-folding methods for both proteins & small molecules, diffusion and non-diffusion-based, to extend on AF2 success: DiffusionProteinLigand in the last days of 2022 and RFDiffusion, AlphaFold2 and Umol by end of 2023. We are also seeing models that have sequence & structure co-trained: SAProt, ProstT5, and sequence, structure & surface co-trained with ProteinINR. There is a general revival of surface-based methods after a quieter 2021 and 2022: DiffMasif, SurfDock, and ShapeProt.
2️⃣ Datasets and benchmarks. Datasets, especially synthetic/computationally derived: ATLAS and the MDDB for protein dynamics. MISATO, SPICE, Splinter for protein-ligand complexes, QM1B for molecular properties. PINDER: large protein-protein docking dataset with matched apo/predicted pairs and benchmark suite with retrained docking models. CryoET data portal for CryoET. And a whole host of welcome benchmarks: PINDER, PoseBusters, and PoseCheck, with a focus on more rigorous and practically relevant settings.
3️⃣ Creative pre-training strategies to get around the sparsity of diverse protein-ligand complexes. Van-der-mers training (DockGen) & sidechain training strategies in RF-AA and pre-training on ligand-only complexes in CCD in RF-AA. Multi-task pre-training Unimol and others.
🏋️ What are the open challenges that researchers might overlook?
1️⃣ Generalization. DockGen showed that current state-of-the-art protein-ligand docking models completely lose predictability when asked to generalise towards novel protein domains. We see a similar phenomenon in the AlphaFold-lastest report, where performance on novel proteins & ligands drops heavily to below biophysics-based baselines (which have access to holo structures), despite very generous definitions of novel protein & ligand. This indicates that existing approaches might still largely rely on memorization, an observation that has been extensively argued over the years
2️⃣ The curse of (simple) baselines. A recurring topic over the years, 2023 has again shown what industry practitioners have long known: in many practical problems such as molecular generation, property prediction, docking, and conformer prediction, simple baselines or classical approaches often still outperform ML-based approaches in practice. This has been documented increasingly in 2023 by Tripp et al., Yu et al., Zhou et al.
🔮 Predictions for 2024!
“In 2024, data sparsity will remain top of mind and we will see a lot of smart ways to use models to generate synthetic training data. Self-distillation in AlphaFold2 served as a big inspiration, Confidence Bootstrapping in DockGen, leveraging the insight that we now have sufficiently powerful models that can score poses but not always generate them, first realised in 2022.” — Luca Naef (VantAI)
2️⃣ We will see more biological/chemical assays purpose-built for ML or only making sense in a machine learning context (i.e., might not lead to biological insight by themselves but be primarily useful for training models). An example from 2023 is the large-scale protein folding experiments by Tsuboyama et al. This move might be driven by techbio startups, where we have seen the first foundation models built on such ML-purpose-built assays for structural biology with e.g. ATOM-1.
Andreas Loukas (Prescient Design, part of Genentech)
🔥 What are the biggest advancements in the field you noticed in 2023?
“In 2023, we started to see some of the challenges of equivariant generation and representation for proteins to be resolved through diffusion models.” — Andreas Loukas (Prescient Design)
1️⃣ We also noticed a shift towards approaches that model and generate molecular systems at higher fidelity. For instance, the most recent models adopt a fully end-to-end approach by generating backbone, sequence and side-chains jointly (AbDiffuser, dyMEAN) or at least solve the problem in two steps but with a partially joint model (Chroma); as compared to backbone generation followed by inverse folding as in RFDiffusion and FrameDiff. Other attempts to improve the modelling fidelity can be found in the latest updates to co-folding tools like AlphaFold2 and RFDiffusion which render them sensitive to non-protein components (ligands, prosthetic groups, cofactors); as well as in papers that attempt to account for conformational dynamics (see discussion above). In my view, this line of work is essential because the binding behaviour of molecular systems can be very sensitive to how atoms are placed, move, and interact.
2️⃣ In 2023, many works also attempted to get a handle on binding affinity by learning to predict the effect of mutations of a known crystal by pre-training on large corpora, such as computationally predicted mutations (graphinity), and on side-tasks, such as rotamer density estimation. The obtained results are encouraging as they can significantly outperform semi-empirical baselines like Rosetta and FoldX. However, there is still significant work to be done to render these models reliable for binding affinity prediction.
3️⃣ I have further observed a growing recognition of protein Language Models (pLMs) and specifically ESM as valuable tools, even among those who primarily favour geometric deep learning. These embeddings are used to help docking models, allow the construction of simple yet competitive predictive models for binding affinity prediction (Li et al 2023), and can generally offer an efficient method to create residue representations for GNNs that are informed by the extensive proteome data without the need for extensive pretraining (Jamasb et al 2023). However, I do maintain a concern regarding the use of pLMs: it is unclear whether their effectiveness is due to data leakage or genuine generalisation. This is particularly pertinent when evaluating models on tasks like amino-acid recovery in inverse folding and conditional CDR design, where distinguishing between these two factors is crucial.
🏋️ What are the open challenges that researchers might overlook?
1️⃣ Working with energetically relaxed crystal structures (and, even worse, folded structures) can significantly affect the performance of downstream predictive models. This is especially true for the prediction of protein-protein interactions (PPIs). In my experience, the performance of PPI predictors severely deteriorates when they are given a relaxed structure as opposed to the binding (holo) crystalised structure.
2️⃣ Though successful in silico antibody design has the capacity to revolutionise drug design, general protein models are not (yet?) as good at folding, docking or generating antibodies as antibody-specific models are. This is perhaps due to the low conformational variability of the antibody fold and the distinct binding mode between antibodies and antigens (loop-mediated interactions that can involve a non-negligible entropic component). Perhaps for the same reasons, the de novo design of antibody binders (that I define as 0-shot generation of an antibody that binds to a previously unseen epitope) remains an open problem. Currently, experimentally confirmed cases of de novo binders involve mostly stable proteins, like alpha-helical bundles, that are common in the PDB and harbour interfaces that differ substantially from epitope-paratope interactions.
3️⃣ We are still lacking a general-purpose proxy for binding free energy. The main issue here is the lack of high-quality data of sufficient size and diversity (esp. co-crystal structures). We should therefore be cognizant of the limitations of any such learned proxy for any model evaluation: though predicted binding scores that are out of distribution of known binders is a clear signal that something is off, we should avoid the typical pitfall of trying to demonstrate the superiority of our model in an empirical evaluation by showing how it leads to even higher scores.
Dominique Beaini (Valence Labs, part of Recursion)
“I’m excited to see a very large community being built around the problem of drug discovery, and I feel we are on the brink of a new revolution in the speed and efficiency of discovering drugs.” — Dominique Beaini (Valence Labs)
What work got me excited in 2023?
I am confident that machine learning will allow us to tackle rare diseases quickly, stop the next COVID-X pandemic before it can spread, and live longer and healthier. But there’s a lot of work to be done and there are a lot of challenges ahead, some bumps in the road, and some canyons on the way. Speaking of communities, you can visit the Valence Portal to keep up-to-date with the 🔥 new in ML for drug discovery.
What are the hard questions for 2024?
⚛️ A new generation of quantum mechanics. Machine learning force-fields, often based on equivariant and invariant GNNs, have been promising us a treasure. The treasure of the precision of density functional theory, but thousands of times faster and at the scale of entire proteins. Although some steps were made in this direction with Allegro and MACE-MP, current models do not generalize well to unseen settings and very large molecules, and they are still too slow to be applicable on the timescale that is needed 🐢. For the generalization, I believe that bigger and more diverse datasets are the most important stepping stones. For the computation time, I believe we will see models that are less enforcing of the equivariance, such as FAENet. But efficient sampling methods will play a bigger role: spatial-sampling such as using DiffDock to get more interesting starting points and time-sampling such as TimeWarp to avoid simulating every frame. I’m really excited by the big STEBS 👣 awaiting us in 2024: Spatio-temporal equivariant Boltzmann samplers.
🕸️ Everything is connected. Biology is inherently multimodal 🙋🐁 🧫🧬🧪. One cannot simply decouple the molecule from the rest of the biological system. Of course, that’s how ML for drug discovery was done in the past: simply build a model of the molecular graph and fit it to experimental data. But we have reached a critical point 🛑, no matter how many trillion parameters are in the GNN model is, and how much data are used to train it, and how many experts are mixtured together. It is time to bring biology into the mix, and the most straightforward way is with multi-modal models. One method is to condition the output of the GNNs with the target protein sequences such as MocFormer. Another is to use microscopy images or transcriptomics to better inform the model of the biological signature of molecules such as TranSiGen. Yet another is to use LLMs to embed contextual information about the tasks such as TwinBooster. Or even better, combining all of these together 🤯, but this could take years. The main issue for the broader community seems to be the availability of large amounts of quality and standardized data, but fortunately, this is not an issue for Valence.
🔬 Relating biological knowledge and observables. Humans have been trying to map biology for a long time, building relational maps for genes 🧬, protein-protein interactions 🔄, metabolic pathways 🔀, etc. I invite you to read this review of knowledge graphs for drug discovery. But all this knowledge often sits unused and ignored by the ML community. I feel that this is an area where GNNs for knowledge graphs could prove very useful, especially in 2024, and it could provide another modality for the 🕸️ point above. Considering that human knowledge is incomplete, we can instead recover relational maps from foundational models. This is the route taken by Phenom1 when trying to recall known genetic relationships. However, having to deal with various knowledge databases is an extremely complex task that we can’t expect most ML scientists to be able to tackle alone. But with the help of artificial assistants like LOWE, this can be done in a matter of seconds.
🏆 Benchmarks, benchmarks, benchmarks. I can’t repeat the word benchmark enough. Alas, benchmarks will stay the unloved kid on the ML block 🫥. But if the word benchmark is uncool, its cousin competition is way cooler 😎! Just as the OGB-LSC competition and Open Catalyst challenge played a major role for the GNN community, it is now time for a new series of competitions 🥇. We even got the TGB (Temporal graph benchmark) recently. If you were at NeurIPS’23, then you probably heard of Polaris coming up early 2024 ✨. Polaris is a consortium of multiple pharma and academic groups trying to improve the quality of available molecular benchmarks to better represent real drug discovery. Perhaps we’ll even see a benchmark suitable for molecular graph generation instead of optimizing QED and cLogP, but I wouldn’t hold my breath, I have been waiting for years. What kind of new, crazy competition will light up the GDL community this year 🤔?
Systems Biology
PINNACLE has protein-, cell type-, and tissue-level attention mechanisms that enable the algorithm to generate contextualized representations of proteins, cell types, and tissues in a single unified embedding space. Source: Li et al
3️⃣ GNNs also have shown a vital role in diagnosing rare diseases. SHEPHERD utilizes GNN over massive knowledge graph to encode extensive biological knowledge into the ML model and is shown to facilitate causal gene discovery, identify ‘patients-like-me’ with similar genes or diseases, and provide interpretable insights into novel disease manifestations.
➡️ Moving beyond predictions, understanding the underlying mechanisms of biological phenomena is crucial. Graph XAI applied to system graphs is a natural fit for identifying mechanistic pathways. TxGNN, for example, grounds drug-disease relation predictions in the biological system graph, generating multi-hop interpretable paths. These paths rationalize the potential of a drug in treating a specific disease. TxGNN designed visualizations for these interpretations and conducted user studies, proving their decision-making effectiveness for clinicians and biomedical scientists.A web-based graphical user interface to support clinicians and scientists in exploring and analyzing the predictions and explanations generated by TxGNN. The ‘Control Panel‘ allows users to select the disease of interest and view the top-ranked TXGNN predictions for the query disease. The ‘edge threshold‘ module enables users to modify the sparsity of the explanation and thereby control the density of the multi-hop paths displayed. The ‘Drug Embedding‘ panel allows users to compare the position of a selected drug relative to the entire repurposing candidate library. The ‘Path Explanation‘ panel displays the biological relations that have been identified as crucial for TXGNN’s predictions regarding therapeutic use. Source: Huang, Chandar, et al
➡️ Foundation models in biology have predominantly been unimodal (focused on proteins, molecules, diseases, etc.), primarily due to the scarcity of paired data. Bridging across modalities to answer multi-modal queries is an exciting frontier. For example, BioBridge leverages biological knowledge graphs to learn transformations across unimodal foundation models, enabling multi-modal behaviors.
🔮 GNNs applied to system graphs have the potential to (1) encode vast biomedical knowledge, (2) bridge biological modalities, (3) provide mechanistic insights, and (4) contextualize biological entities. We anticipate even more groundbreaking applications of GNN in biology in 2024, addressing some of the most pressing questions in the field.
Predictions from the 2023 post
(1) performance improvements of diffusion models such as faster sampling and more efficient solvers;
✅ yes, with flow matching
(2) more powerful conditional protein generation models;
❌ Chroma and RFDiffusion are still on top
(3) more successful applications of Generative Flow Networks to molecules and proteins
❌ yet to be seen
Materials Science (Crystals)
Michael Galkin (Intel) and Santiago Miret (Intel)
In 2023, for a short period, all scientific news were talking only about LK-99 — a supposed room-temperature superconductor created by a Korean team (spoiler: it did not work as of now).
This highlights the huge potential ML has in material science, where perhaps the biggest progress of the year has happened — we can now say that materials science and materials discovery are first-class citizens in the Geometric DL landscape.
💡The advances of Geometric DL applied to materials science and discovery saw significant advances across new modelling methods, creation of new benchmarks and datasets, automated design with generative methods, and identifying new research questions based on those advances.
1️⃣ Applications of geometric models as evaluation tools in automated discovery workflows. The Open MatSci ML Toolkit consolidated all open-sourced crystal structures datasets leading to 1.5 million data points for ground-state structure calculations that are now easily available for model development. The authors’ initial results seem to indicate that merging datasets seems to improve performance if done attentively.
2️⃣ MatBench Discovery is another good example of this integration of geometric models as an evaluation tool for crystal stability, which tests models’ predictions of the energy above hull for various crystal structures. The energy above hull is the most reliable approximation of crystal structure stability and also represents an improvement in metrics compared to formation energy or raw energy prediction which have practical limitations as stability metrics.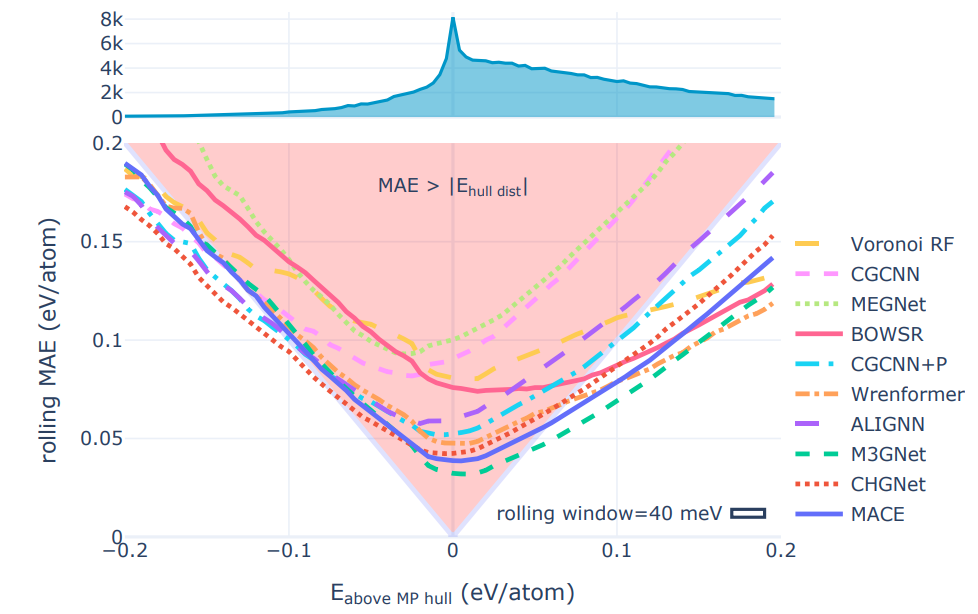 Universal potentials are more reliable classifiers because they exit the red triangle earliest. These lines show the rolling MAE on the WBM test set as the energy to the convex hull of the MP training set is varied, lower is better. The red-highlighted ’triangle of peril’ shows where the models are most likely to misclassify structures. As long as a model’s rolling MAE remains inside the triangle, its mean error is larger than the distance to the convex hull. If the model’s error for a given prediction happens to point towards the stability threshold at 0 eV from the hull (the plot’s center), its average error will change the stability classification of a material from true positive/negative to false negative/positive. The width of the ’rolling window’ box indicates the width over which errors hull distance prediction errors were averaged. Source: Riebesell et al
Universal potentials are more reliable classifiers because they exit the red triangle earliest. These lines show the rolling MAE on the WBM test set as the energy to the convex hull of the MP training set is varied, lower is better. The red-highlighted ’triangle of peril’ shows where the models are most likely to misclassify structures. As long as a model’s rolling MAE remains inside the triangle, its mean error is larger than the distance to the convex hull. If the model’s error for a given prediction happens to point towards the stability threshold at 0 eV from the hull (the plot’s center), its average error will change the stability classification of a material from true positive/negative to false negative/positive. The width of the ’rolling window’ box indicates the width over which errors hull distance prediction errors were averaged. Source: Riebesell et al
3️⃣ In terms of new geometric models for crystal structure prediction, Crystal Hamiltonian Graph neural network (CHGNet, Deng et al) is a new GNN trained on static and relaxation trajectories of Materials Project that shows quite competitive performance compared to prior methods. The development of CHGNet suggests that finding better training objectives will be as (if not more) important than the development of new methods as the intersection of materials science and geometric deep learning continues to grow.
🔥 The other proof points of the further integration of Geometric DL and materials discovery are several massive works by big labs focused on crystal structure discovery with generative methods:
1️⃣ Google DeepMind released GNoME (Graph Networks for Materials Science by Merchant et al) as a successful example of an active learning pipeline for discovering new materials, and UniMat as an ab initio crystal generation model. Similar to the protein world, we see more examples of automated labs for materials science (“lab-in-the-loop”) such as the A-Lab from UC Berkley.The active learning loop of GNoME. Source: Merchant et al.
2️⃣ Microsoft Research released MatterGen, a generative model for unconditional and property-guided materials design, and Distributional Graphormer, a generative model trained to recover the equilibrium energy distribution of a molecule/protein/crystal.Unconditional and conditional generation of MatterGen. Source: Zeni, Pinsler, Zügner, Fowler, Horton, et al.
3️⃣ Meta AI and CMU released the Open Catalyst Demo where you can play around with relaxations (DFT approximations) of 11.5k catalyst materials on 86 adsorbates in 100 different configurations each (making it up to 100M combinations). The demo is powered by SOTA geometric models GemNet-OC and Equiformer-V2.
This article owner is :here Im just share this article because i like AI